
Early-Stage Lung Adenocarcinoma MDM2 Genomic Amplification Predicts Clinical Outcome and Response to Targeted Therapy

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. SNP Array
2.2. Chromogenic In Situ Hybridization (CISH) and Fluorescent In Situ Hybridization (FISH)
2.3. Patient Data Analysis for MDM2 and p53 Heterogeneity Distribution
2.4. Immunohistochemistry
2.5. Survival Analysis
2.6. Cell Culture
2.7. MDM2 Inhibitor
2.8. Migration and Invasion
2.9. Wound Healing Assay
2.10. Western Blot
2.11. Proliferation Assay
2.12. RNA Sequencing and Library Preparation
2.13. RNA-Seq Analysis
2.14. Gene Set Enrichment Analysis
2.15. Statistical Analysis
3. Results
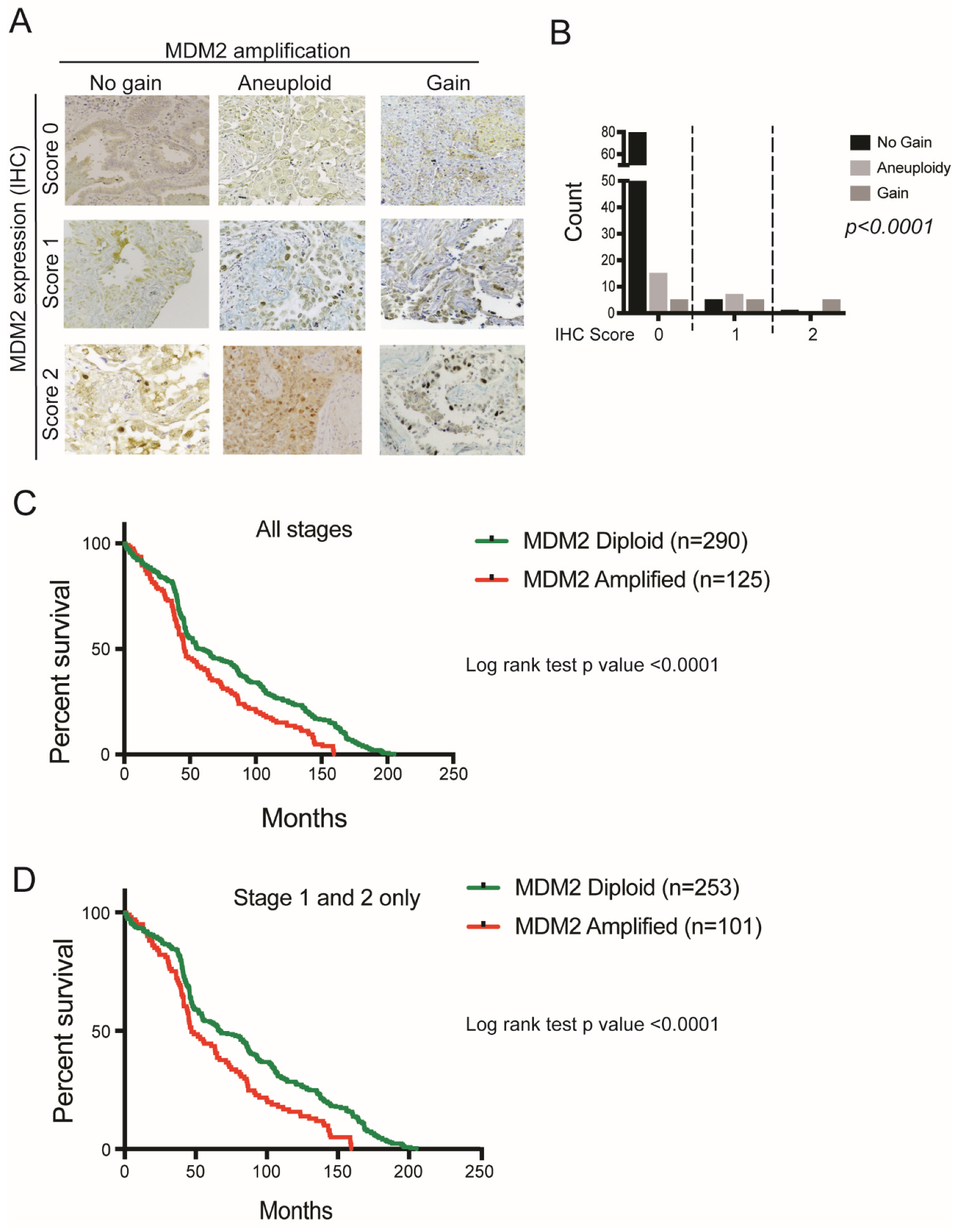
3.1. MDM2 Genomic Heterogeneity in LUAD Patients
3.2. MDM2 Genomic Amplification Is Associated with Protein Overexpression and Poor Survival in LUAD
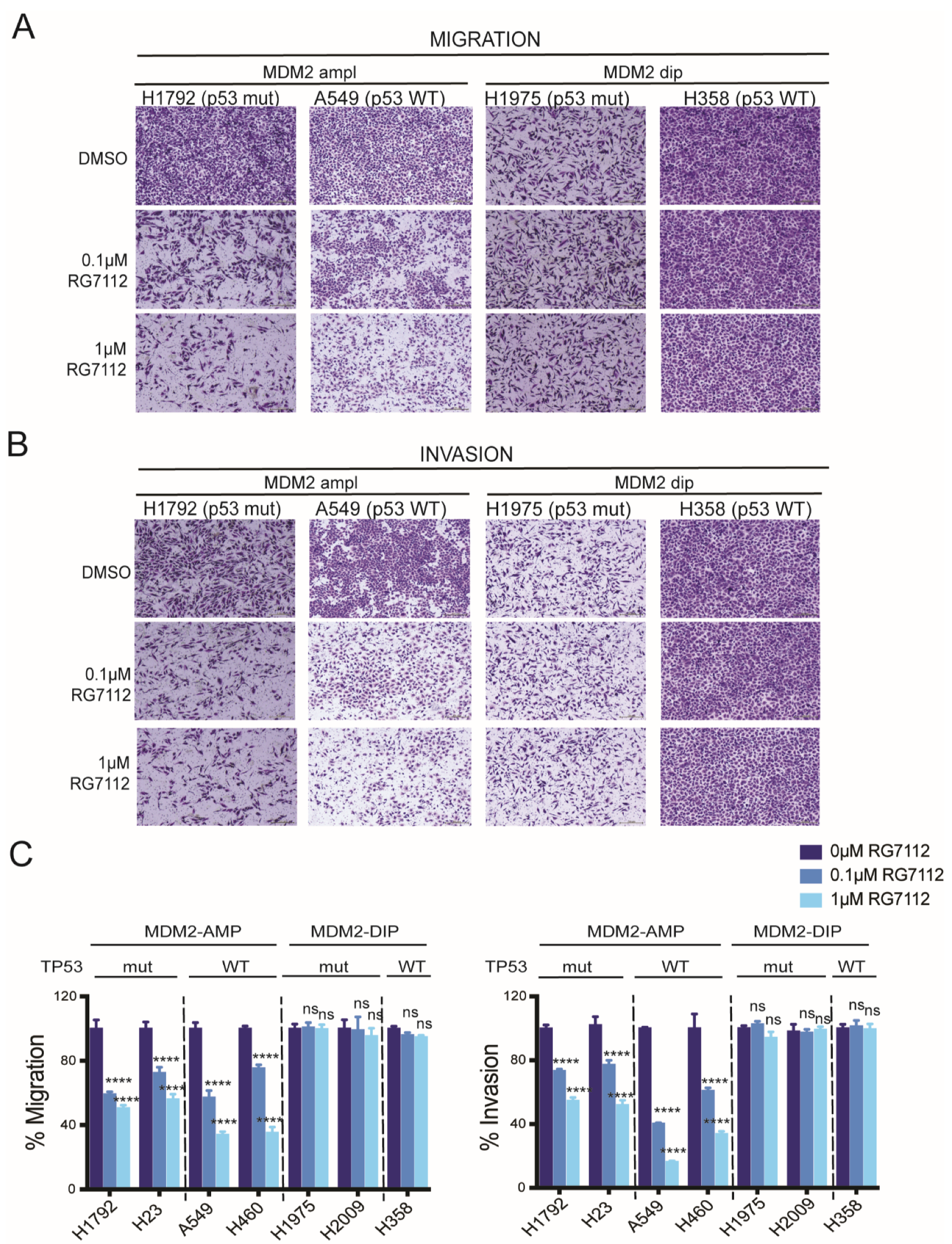
3.3. Anti-Migratory and Anti-Invasive Effect of MDM2 Inhibitor in MDM2-Amplified Tumor Cells
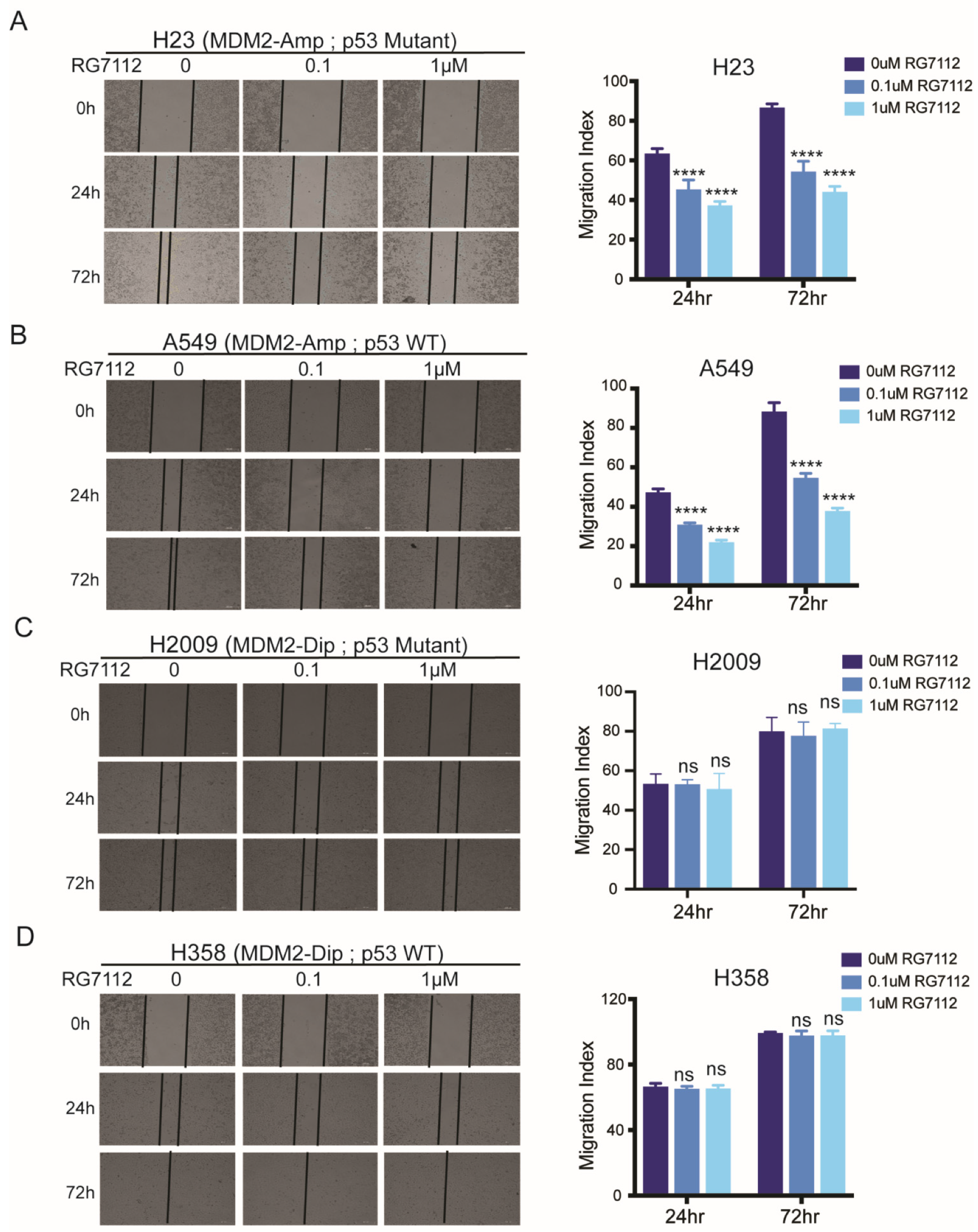
3.4. MDM2 Inhibitor Suppresses Cell Motility in MDM2 Dependent Manner
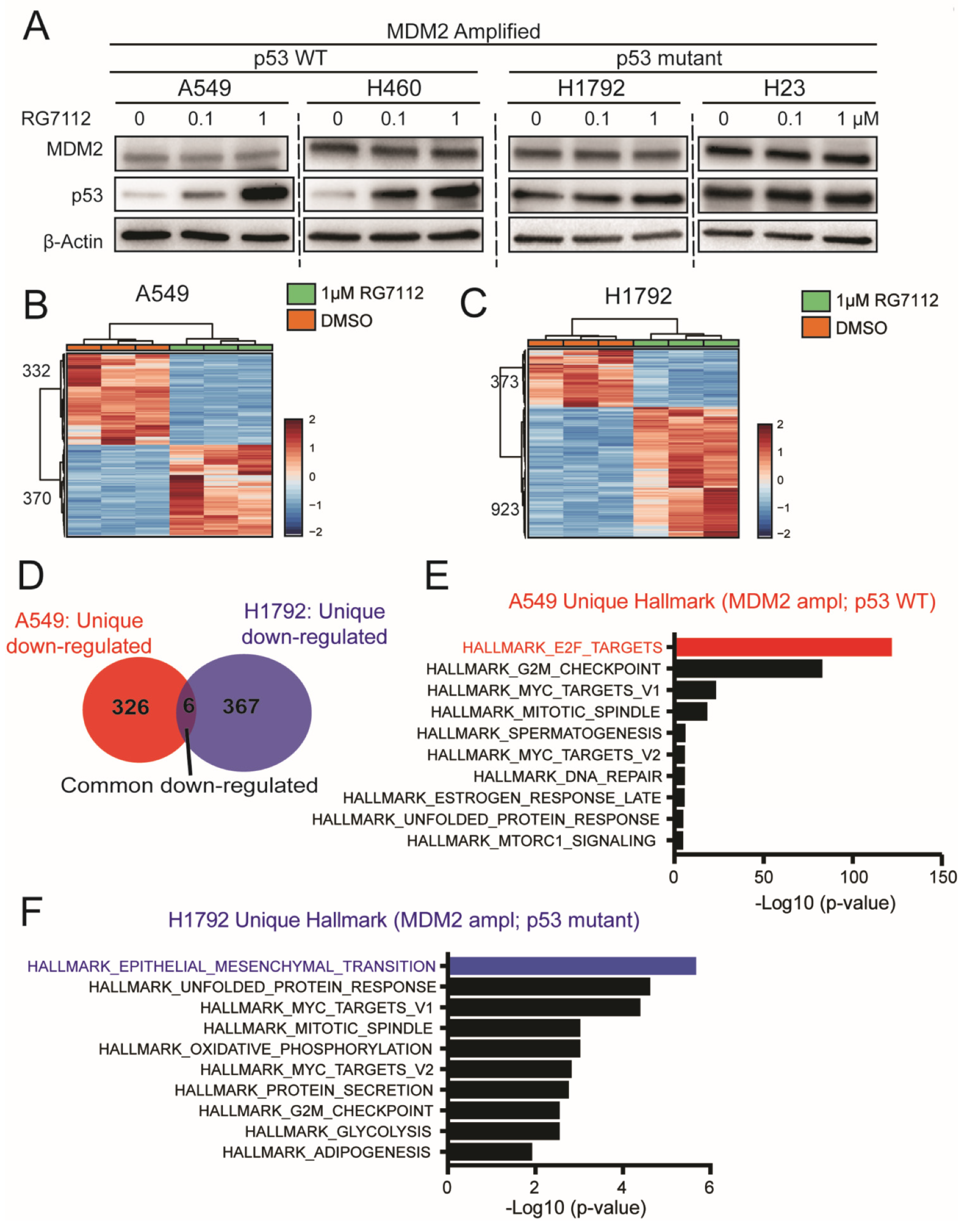
3.5. MDM2 Inhibitor Alters Unique Signaling in MDM2 Amplified; p53 Mutant vs. WT Background
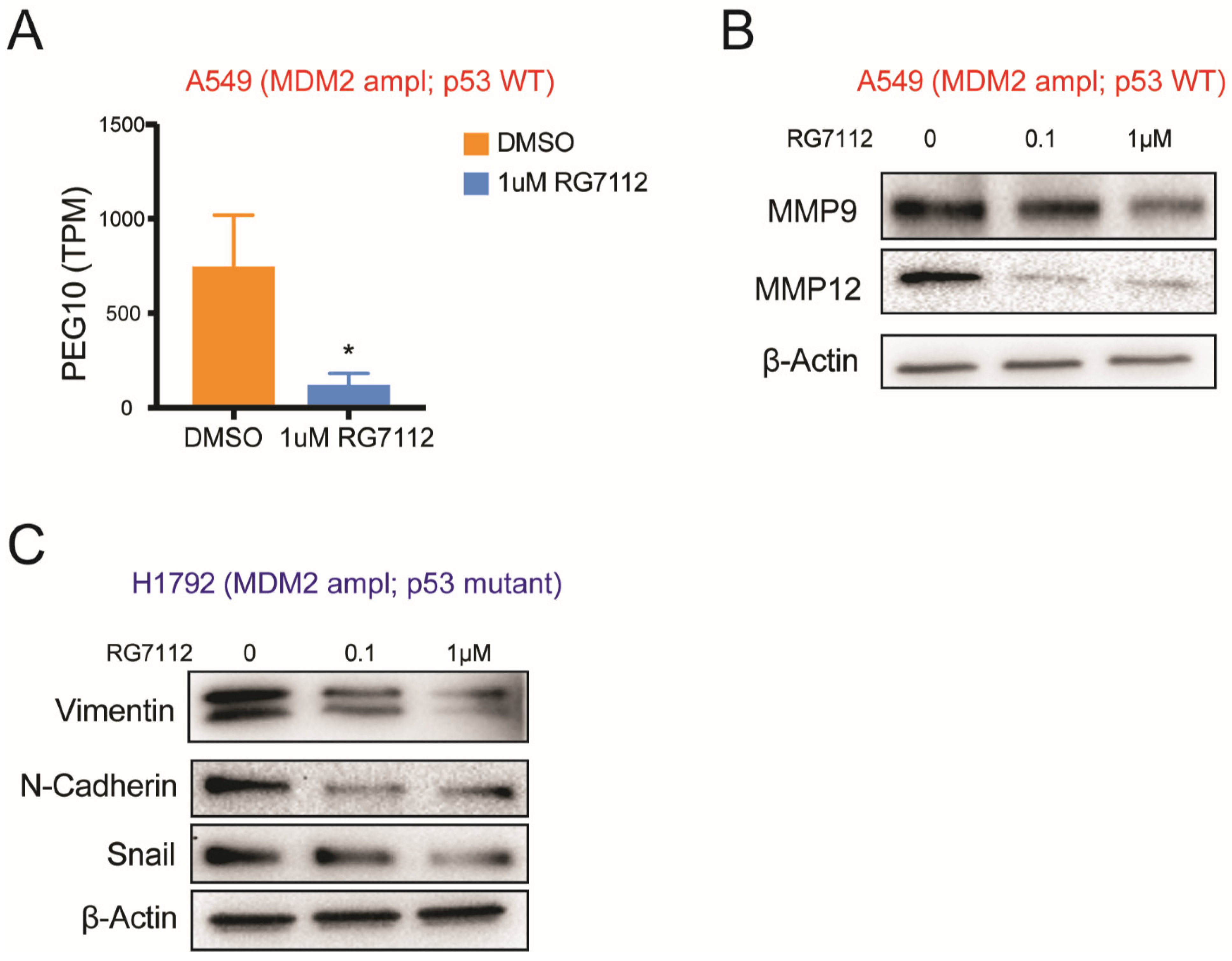
3.6. MDM2-Targeted Therapy Suppresses Tumorigenesis in p53 Wild-Type Condition through E2F → PEG10 → MMP Signaling
3.7. MDM2 Inhibitor Suppresses Invasiveness in MDM2 Amplified; p53 Mutant Group through Suppressing EMT Signaling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jamal-Hanjani, M.; Hackshaw, A.; Ngai, Y.; Shaw, J.; Dive, C.; Quezada, S.; Middleton, G.; de Bruin, E.; Le Quesne, J.; Shafi, S.; et al. Tracking Genomic Cancer Evolution for Precision Medicine: The Lung TRACERx Study. PLoS Biol. 2014, 12, e1001906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Yu, H.; Hu, W. The Regulation of MDM2 Oncogene and Its Impact on Human Cancers. Acta Biochim. Biophys. Sin. 2014, 46, 180–189. [Google Scholar] [CrossRef] [Green Version]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-P53 Pathway Revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic Mutations Affect Key Pathways in Lung Adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature 2014, 511, 543–550. [CrossRef] [PubMed]
- Qian, J.; Zhao, S.; Zou, Y.; Rahman, S.M.J.; Senosain, M.-F.; Stricker, T.; Chen, H.; Powell, C.A.; Borczuk, A.C.; Massion, P.P. Genomic Underpinnings of Tumor Behavior in In Situ and Early Lung Adenocarcinoma. Am. J. Respir. Crit. Care Med. 2020, 201, 697–706. [Google Scholar] [CrossRef]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the Hallmarks of Lung Adenocarcinoma with Massively Parallel Sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-Optimal Probabilistic RNA-Seq Quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Cha, M.J.; Lee, H.Y.; Lee, K.S.; Jeong, J.Y.; Han, J.; Shim, Y.M.; Hwang, H.S. Micropapillary and Solid Subtypes of Invasive Lung Adenocarcinoma: Clinical Predictors of Histopathology and Outcome. J. Thorac. Cardiovasc. Surg. 2014, 147, 921–928.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, P.A.; Wainer, Z.; Wright, G.M.; Daniels, M.; Conron, M.; Williams, R.A. Does Lung Adenocarcinoma Subtype Predict Patient Survival?: A Clinicopathologic Study Based on the New International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society International Multidisciplinary Lung Adenocarcinoma Classification. J. Thorac. Oncol. 2011, 6, 1496–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Tavora, F.; Burke, A. Histologic Features Associated with Metastatic Potential in Invasive Adenocarcinomas of the Lung. Am. J. Surg. Pathol. 2013, 37, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wang, R.; Han, B.; Zhang, J.; Zhao, H.; Fang, W.; Luo, Q.; Yang, J.; Yang, Y.; Zhu, L.; et al. Solid Predominant Histologic Subtype and Early Recurrence Predict Poor Postrecurrence Survival in Patients with Stage I Lung Adenocarcinoma. Oncotarget 2017, 8, 7050–7058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, B.; Wovkulich, P.; Pizzolato, G.; Lovey, A.; Ding, Q.; Jiang, N.; Liu, J.-J.; Zhao, C.; Glenn, K.; Wen, Y.; et al. Discovery of RG7112: A Small-Molecule MDM2 Inhibitor in Clinical Development. ACS Med. Chem. Lett. 2013, 4, 466–469. [Google Scholar] [CrossRef] [Green Version]
- Sadasivam, S.; DeCaprio, J.A. The DREAM Complex: Master Coordinator of Cell Cycle-Dependent Gene Expression. Nat. Rev. Cancer 2013, 13, 585–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roveimiab, Z.; Lin, F.; Anderson, J.E. Traction and Attraction: Haptotaxis Substrates Collagen and Fibronectin Interact with Chemotaxis by HGF to Regulate Myoblast Migration in a Microfluidic Device. Am. J. Physiol. -Cell Physiol. 2020, 319, C75–C92. [Google Scholar] [CrossRef]
- Haeger, A.; Wolf, K.; Zegers, M.M.; Friedl, P. Collective Cell Migration: Guidance Principles and Hierarchies. Trends Cell Biol. 2015, 25, 556–566. [Google Scholar] [CrossRef]
- Park, J.S.; Rhau, B.; Hermann, A.; McNally, K.A.; Zhou, C.; Gong, D.; Weiner, O.D.; Conklin, B.R.; Onuffer, J.; Lim, W.A. Synthetic Control of Mammalian-Cell Motility by Engineering Chemotaxis to an Orthogonal Bioinert Chemical Signal. Proc. Natl. Acad. Sci. USA 2014, 111, 5896–5901. [Google Scholar] [CrossRef] [Green Version]
- Kölsch, V.; Charest, P.G.; Firtel, R.A. The Regulation of Cell Motility and Chemotaxis by Phospholipid Signaling. J. Cell Sci. 2008, 121, 551–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Tonqueze, O.; Kollu, S.; Lee, S.; Al-Salah, M.; Truesdell, S.S.; Vasudevan, S. Regulation of Monocyte Induced Cell Migration by the RNA Binding Protein, FXR1. Cell Cycle 2016, 15, 1874–1882. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Hassanein, M.; Hoeksema, M.D.; Harris, B.K.; Zou, Y.; Chen, H.; Lu, P.; Eisenberg, R.; Wang, J.; Espinosa, A.; et al. The RNA Binding Protein FXR1 Is a New Driver in the 3q26-29 Amplicon and Predicts Poor Prognosis in Human Cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 3469–3474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, S.; Zheng, J.; Liu, X.; Liu, Y.; Ruan, X.; Ma, J.; Liu, L.; Wang, D.; Yang, C.; Cai, H.; et al. FXR1 Promotes the Malignant Biological Behavior of Glioma Cells via Stabilizing MIR17HG. J. Exp. Clin. Cancer Res. 2019, 38, 37. [Google Scholar] [CrossRef]
- Zhou, L.; Panté, N. The Nucleoporin Nup153 Maintains Nuclear Envelope Architecture and Is Required for Cell Migration in Tumor Cells. FEBS Lett. 2010, 584, 3013–3020. [Google Scholar] [CrossRef] [Green Version]
- Chow, K.-H.; Factor, R.E.; Ullman, K.S. The Nuclear Envelope Environment and Its Cancer Connections. Nat. Rev. Cancer 2012, 12, 196–209. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Song, Q.; Zeng, R.; Li, J.; Li, J.; Lin, X.; Chen, X.; Zhang, J.; Zheng, Y. MicroRNA-218 Inhibits EMT, Migration and Invasion by Targeting SFMBT1 and DCUN1D1 in Cervical Cancer. Oncotarget 2016, 7, 45622–45636. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.; Shen, H.; Jin, Y.; Lin, T.; Cai, Q.; Pinard, M.A.; Biswas, S.; Tran, Q.; Li, G.; Shenoy, A.K.; et al. The Malignant Brain Tumor (MBT) Domain Protein SFMBT1 Is an Integral Histone Reader Subunit of the LSD1 Demethylase Complex for Chromatin Association and Epithelial-to-Mesenchymal Transition. J. Biol. Chem. 2013, 288, 27680–27691. [Google Scholar] [CrossRef] [Green Version]
- Forma, E.; Jóźwiak, P.; Bryś, M.; Krześlak, A. The Potential Role of O-GlcNAc Modification in Cancer Epigenetics. Cell. Mol. Biol. Lett. 2014, 19, 438–460. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.-P.; Zhu, Y.; Yin, L.-D.; Zhang, J.-J.; Wei, J.-S.; Liu, X.; Liu, X.-C.; Gao, W.-T.; Jiang, K.-R.; Miao, Y. PEG10 Overexpression Induced by E2F-1 Promotes Cell Proliferation, Migration, and Invasion in Pancreatic Cancer. J. Exp. Clin. Cancer Res. 2017, 36, 30. [Google Scholar] [CrossRef] [Green Version]
- Xie, T.; Pan, S.; Zheng, H.; Luo, Z.; Tembo, K.M.; Jamal, M.; Yu, Z.; Yu, Y.; Xia, J.; Yin, Q.; et al. PEG10 as an Oncogene: Expression Regulatory Mechanisms and Role in Tumor Progression. Cancer Cell Int. 2018, 18, 112. [Google Scholar] [CrossRef] [PubMed]
- Akamatsu, S.; Wyatt, A.W.; Lin, D.; Lysakowski, S.; Zhang, F.; Kim, S.; Tse, C.; Wang, K.; Mo, F.; Haegert, A.; et al. The Placental Gene PEG10 Promotes Progression of Neuroendocrine Prostate Cancer. Cell Rep. 2015, 12, 922–936. [Google Scholar] [CrossRef] [Green Version]
- Kato, S.; Ross, J.S.; Gay, L.; Dayyani, F.; Roszik, J.; Subbiah, V.; Kurzrock, R. Analysis of MDM2 Amplification: Next-Generation Sequencing of Patients with Diverse Malignancies. JCO Precis. Oncol. 2018, 2018, 1–14. [Google Scholar] [CrossRef]
- Sciot, R. MDM2 Amplified Sarcomas: A Literature Review. Diagnostics 2021, 11, 496. [Google Scholar] [CrossRef] [PubMed]
- Günther, T.; Schneider-Stock, R.; Häckel, C.; Kasper, H.-U.; Pross, M.; Hackelsberger, A.; Lippert, H.; Roessner, A. Mdm2 Gene Amplification in Gastric Cancer Correlation with Expression of Mdm2 Protein and P53 Alterations. Mod. Pathol. 2000, 13, 621–626. [Google Scholar] [CrossRef]
- Michalk, M.; Meinrath, J.; Künstlinger, H.; Koitzsch, U.; Drebber, U.; Merkelbach-Bruse, S.; Bollschweiler, E.; Kloth, M.; Hartmann, W.; Hölscher, A.; et al. MDM2 Gene Amplification in Esophageal Carcinoma. Oncol. Rep. 2016, 35, 2223–2227. [Google Scholar] [CrossRef] [Green Version]
- Oliner, J.D.; Saiki, A.Y.; Caenepeel, S. The Role of MDM2 Amplification and Overexpression in Tumorigenesis. Cold Spring Harb. Perspect. Med. 2016, 6, a026336. [Google Scholar] [CrossRef]
- Reis, B.; Jukofsky, L.; Chen, G.; Martinelli, G.; Zhong, H.; So, W.V.; Dickinson, M.J.; Drummond, M.; Assouline, S.; Hashemyan, M.; et al. Acute Myeloid Leukemia Patients Clinical Response to Idasanutlin (RG7388) Is Associated with Pre-Treatment MDM2 Protein Expression in Leukemic Blasts. Haematologica 2016, 101, e185–e188. [Google Scholar] [CrossRef] [Green Version]
- Montesinos, P.; Beckermann, B.M.; Catalani, O.; Esteve, J.; Gamel, K.; Konopleva, M.Y.; Martinelli, G.; Monnet, A.; Papayannidis, C.; Park, A.; et al. MIRROS: A Randomized, Placebo-Controlled, Phase III Trial of Cytarabine ± Idasanutlin in Relapsed or Refractory Acute Myeloid Leukemia. Future Oncol. 2020, 16, 807–815. [Google Scholar] [CrossRef]
- Sun, D.; Li, Z.; Rew, Y.; Gribble, M.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chen, X.; Chow, D.; et al. Discovery of AMG 232, a Potent, Selective, and Orally Bioavailable MDM2–P53 Inhibitor in Clinical Development. J. Med. Chem. 2014, 57, 1454–1472. [Google Scholar] [CrossRef]
- Zito Marino, F.; Bianco, R.; Accardo, M.; Ronchi, A.; Cozzolino, I.; Morgillo, F.; Rossi, G.; Franco, R. Molecular Heterogeneity in Lung Cancer: From Mechanisms of Origin to Clinical Implications. Int. J. Med. Sci. 2019, 16, 981–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinha, A.; Zou, Y.; Patel, A.S.; Yoo, S.; Jiang, F.; Sato, T.; Kong, R.; Watanabe, H.; Zhu, J.; Massion, P.P.; et al. Early-Stage Lung Adenocarcinoma MDM2 Genomic Amplification Predicts Clinical Outcome and Response to Targeted Therapy. Cancers 2022, 14, 708. https://doi.org/10.3390/cancers14030708
Sinha A, Zou Y, Patel AS, Yoo S, Jiang F, Sato T, Kong R, Watanabe H, Zhu J, Massion PP, et al. Early-Stage Lung Adenocarcinoma MDM2 Genomic Amplification Predicts Clinical Outcome and Response to Targeted Therapy. Cancers. 2022; 14(3):708. https://doi.org/10.3390/cancers14030708
Chicago/Turabian StyleSinha, Abhilasha, Yong Zou, Ayushi S. Patel, Seungyeul Yoo, Feng Jiang, Takashi Sato, Ranran Kong, Hideo Watanabe, Jun Zhu, Pierre P. Massion, and et al. 2022. "Early-Stage Lung Adenocarcinoma MDM2 Genomic Amplification Predicts Clinical Outcome and Response to Targeted Therapy" Cancers 14, no. 3: 708. https://doi.org/10.3390/cancers14030708